
Болезнь менкеса что это
Опубликовано: 17.09.2024
Болезнь Менкеса ( MNK ), также известная как синдром Менкеса , представляет собой Х-сцепленное рецессивное заболевание, вызванное мутациями в генах, кодирующих белок -транспортник меди ATP7A , что приводит к дефициту меди . Характерные признаки включают курчавые волосы, задержку роста и ухудшение нервной системы. Как и все Х-сцепленные рецессивные состояния, болезнь Менкеса чаще встречается у мужчин, чем у женщин. Расстройство было впервые описано Джоном Гансом Менкесом в 1962 году.
Начало происходит в младенчестве, с заболеваемостью примерно от 1 на 100 000 до 250 000 новорожденных; пораженные младенцы часто не доживают до трех лет, хотя в редких случаях менее серьезные симптомы проявляются позже в детстве.
Содержание
- 1 Признаки и симптомы
- 2 Причина
- 3 Механизм
- 4 Диагностика
- 5 Лечение
- 6 Эпидемиология
- 7 См. Также
- 8 ссылки
- 9 Внешние ссылки
Признаки и симптомы
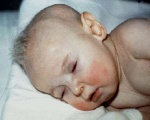
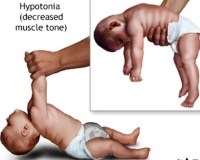
Больные младенцы могут родиться преждевременно . Признаки и симптомы появляются в младенчестве, обычно после двух-трехмесячного периода нормального или слегка замедленного развития, за которым следует утрата навыков раннего развития и последующая задержка в развитии . Пациенты обнаруживают гипотонию (слабый мышечный тонус ), отказ процветать , гипотермию ( субнормальная температуры тела ), провисание черты лица, судороги и метафизарную расширение. Волосы выглядят поразительно своеобразно: курчавые, бесцветные или серебристые и ломкие. Там может быть обширной нейродегенерация в сером веществе в мозге . Артерии головного мозга также могут быть перекручены с потрепанными и раздвоенными внутренними стенками. Это может привести к разрыву или закупорке артерий. Ослабленные кости ( остеопороз ) могут привести к переломам.
Синдром затылочного рога (иногда называемый Х-сцепленной кутис-лаксой или типом Элерса-Данлоса 9) - это легкая форма синдрома Менкеса, которая начинается в раннем и среднем детстве. Он характеризуется отложениями кальция в кости у основания черепа ( затылочная кость ), грубыми волосами, дряблой кожей и суставами.
Причина
Мутации в ATP7A гена , расположенные на хромосоме Xq21.1 , приводит к синдрому Menkes. Это состояние наследуется по Х-сцепленному рецессивному паттерну. Около 30% случаев MNK связаны с новыми мутациями, а 70% наследуются, почти всегда от матери. Несмотря на то, что заболевание чаще встречается у мужчин, женщины все же могут быть носителями болезни. В результате мутации в гене ATP7A медь плохо распределяется по клеткам организма. Медь накапливается в некоторых тканях, таких как тонкий кишечник и почки , в то время как в мозге и других тканях ее уровень необычно низкий. Уменьшение поступления меди может снизить активность многочисленных медьсодержащих ферментов , которые необходимы для структуры и функции костей , кожи , волос , кровеносных сосудов и нервной системы, таких как лизилоксидаза . Как и в случае других Х-сцепленных расстройств, дети женского пола от матери-носителя имеют равные шансы перенести расстройство, но обычно здоровы; Дети мужского пола имеют равные шансы заболеть расстройством или избавиться от него. У генетического консультанта может быть полезный совет.
Механизм

Ген ATP7A кодирует трансмембранный белок, который переносит медь через клеточные мембраны. Он находится по всему телу, кроме печени. В тонком кишечнике белок ATP7A помогает контролировать всасывание меди из пищи. В других клетках белок перемещается между аппаратом Гольджи и клеточной мембраной, чтобы поддерживать концентрацию меди в клетке. Белок обычно находится в аппарате Гольджи, что важно для модификации белков, включая ферменты. В аппарате Гольджи белок ATP7A обеспечивает медью определенные ферменты, которые имеют решающее значение для структуры и функции костей, кожи, волос, кровеносных сосудов и нервной системы. Один из ферментов, лизилоксидаза, требует меди для правильного функционирования. Этот фермент связывает тропоколлаген в прочные фибриллы коллагена. Дефектный коллаген способствует многим из вышеупомянутых проявлений этого заболевания в соединительной ткани.
Если уровень меди становится чрезмерным, белок переместится к клеточной мембране и выведет излишки меди из клетки. Мутации в гене ATP7A, такие как делеции и вставки, приводят к удалению частей гена, что приводит к укорочению белка ATP7A. Это предотвращает выработку функционального белка ATP7A, что приводит к нарушению всасывания меди из пищи, и медь не поступает в определенные ферменты.
Диагностика
Синдром Менкеса можно диагностировать с помощью анализов крови на уровни меди и церулоплазмина, биопсии кожи и оптического микроскопического исследования волос для выявления характерных аномалий Менкеса. Рентген черепа и скелета проводится для выявления аномалий костеобразования. Соотношение гомованилловой кислоты и ваниллилминдальной кислоты в моче было предложено в качестве инструмента скрининга для более раннего обнаружения. Поскольку 70% случаев MNK передаются по наследству, можно провести генетическое тестирование матери для поиска мутации в гене ATP7A.
От болезни Менкеса нет лекарства. Раннее лечение с помощью инъекций добавок меди (ацетата или глицината) может принести небольшую пользу. Среди 12 новорожденных, которым был поставлен диагноз МНК, 92% были живы в возрасте 4,6 года. Другое лечение симптоматическое и поддерживающее. Лечение, которое помогает облегчить некоторые симптомы, включает обезболивающие, противосудорожные препараты, зонд для кормления при необходимости, а также физиотерапию и профессиональную терапию. Чем раньше будет назначено лечение, тем лучше прогноз.
Эпидемиология
Одно европейское исследование сообщило о 1 из 254 000; Японское исследование показало 1 из 357 143. Корреляция с другими унаследованными характеристиками или этническим происхождением неизвестна.
Болезнь Менкеса (болезнь курчавых волос) — это редкое генетическое заболевание, при котором в организме нарушается обмен меди. Патология имеет Х-сцепленный рецессивный тип наследования. Болезнь Менкеса проявляется отставанием в психическом и физическом развитии, ломкостью курчавых волос, церебральными нейродегенеративными поражениями. Диагностика заболевания предполагает генетический анализ, исследование концентрации меди, церулоплазмина, методы нейровизуализации (магнитно-резонансную и компьютерную томографию головного мозга). При болезни курчавых волос проводится заместительная терапия парентеральными препаратами меди, симптоматическое лечение антиконвульсантами и анальгетиками.
МКБ-10
- Причины
- Патогенез
- Симптомы болезни Менкеса
- Осложнения
- Диагностика
- Лечение болезни Менкеса
- Прогноз и профилактика
- Цены на лечение
Общие сведения
Патология относится к орфанным заболеваниям, встречается с частотой от 1:40000 до 1:250000 новорожденных. Болезнь развивается только у мальчиков и названа в честь детского невролога Дж. Х. Менкеса, который описал характерные симптомы в 1962 году. Легким вариантом патологии является синдром затылочного рога, который манифестирует после годовалого возраста и не вызывает летальных осложнений. Его отличительные черты — чрезмерная кальцификация затылочной кости, грубые и жесткие волосы, дряблость кожных покровов.
Причины
Этиологический фактор патологии расшифрован благодаря достижениям медицинской генетики. Болезнь Менкеса возникает при мутации гена полипептид альфа ATP7A, который регулирует работу АТФазы, отвечающей за транспорт ионов меди. Изменение происходит на длинном плече Х-хромосомы в локусе Xq12-q13. Болезнь курчавых волос имеет рецессивное наследование, поэтому поражает только мальчиков. Девочки являются носительницами мутантного гена, но случаев их заболевания не зафиксировано.
Патогенез
При этой наследственной патологии нарушается строение одного из трансмембранных белков, в результате чего ионы меди не могут нормально транспортироваться и участвовать в метаболических процессах. У ребенка возникает дефицит медьсодержащих протеинов, которые в норме используются для обмена веществ, тканевого дыхания, окислительно-восстановительных реакций.
Недостаток микроэлемента вызывает нарушения продукции меланина, уменьшение концентрации коллагена и кератина в тканях, снижение способности к дезинтоксикации свободных радикалов. Важное звено патогенеза — снижение синтеза дофамина, что провоцирует серьезные неврологические расстройства в раннем возрасте. Поскольку медьсодержащий белок церулоплазмин отвечает за окисление железа, при его отсутствии угнетается кроветворение.
Симптомы болезни Менкеса
Болезнь Менкеса может вызывать осложнения беременности — трудные или преждевременные роды, но до 70% женщин не сталкиваются с патологиями интранатального периода. У некоторых младенцев во время родов возникают кефалогематомы и переломы даже при отсутствии активных действий со стороны акушеров. К настораживающим симптомам относят воронкообразную деформацию грудной клетки, врожденную пупочную или паховую грыжу.
Первые признаки болезни курчавых волос появляются в период новорожденности. У младенца наблюдается гипотермия, затяжная желтуха с высокой билирубинемией, удлиняются сроки физиологической послеродовой адаптации. Иногда в неонатальном периоде родители замечают характерные изменения волос: образование узелков и формирование веретенообразных («курчавых») волосков. В этом время физическое и психическое развитие новорожденного соответствует норме.
Явные признаки генетического заболевания регистрируются спустя 2-3 месяца жизни. Становится заметным неврологический дефицит. Ребенок отстает в психомоторном развитии и забывает ранее приобретенные навыки, у него возникают периодические судорожные припадки. На поздних стадиях болезнь проявляется спастическим тетрапарезом вследствие обширных нейродегенеративных церебральных процессов. Кожа приобретает болезненную бледность и желтый оттенок.
На первом году жизни выявляются патогномоничные изменения в виде курчавых волос, обусловленные трихополидистрофией, благодаря которым генетический синдром получил второе название. Волосы становятся тусклыми и жесткими, приобретают серый или «седой» оттенок, закручиваются в тугие спиральки. Нарушения обмена коллагена проявляются чрезмерной растяжимостью кожи и гиперподвижностью в суставах, что клинически напоминает синдром Элерса-Данлоса.
Осложнения
Для синдрома курчавых волос типичны аномалии мочевыделительной системы, особенно дивертикулы мочевого тракта, которые становятся причиной рецидивирующих урогенитальных инфекций. Широко распространены сосудистые аномалии — удлинение, чрезмерная извитость и изменения диаметра артерий. Чаще всего поражаются сосуды головного мозга и кожи. У детей возникает генерализованный остеопороз и спонтанные переломы. Болезнь Менкеса ассоциирована с себорейным дерматитом.
К поздним последствиям заболевания относят слепоту, дыхательную недостаточность. Наиболее тяжело протекают сердечно-сосудистые осложнения. У ребенка могут быть тромботические нарушения, спонтанные разрывы артерий, гематомы. Большинство больных умирают в первые 3 года жизни от генерализованных инфекций, внутричерепных кровоизлияний, грубых нейродегенеративных процессов.
Диагностика
Опытный педиатр может предположить болезнь курчавых волос при физикальном осмотре ребенка, выявлении характерной трихополидистрофии и неврологических нарушений. Постановка диагноза предполагает инструментальную визуализацию для оценки состояния пораженных органов и лабораторные тесты для верификации нарушений обмена меди и генных мутаций. Основные составляющие плана диагностики:
- МРТ головного мозга. Нейровизуализационные исследования показывают очаги кортикальной и субкортикальной атрофии, фокальные участки некроза сером веществе. Типичный симптом болезни Менкеса — атрофия мозжечка. При подозрении на внутричерепное кровоизлияние высокую информативность дает КТ головного мозга.
- ЭЭГ. Исследование производится для выяснения причины судорожных припадков и обнаружения очагов эпилептиформной активности в головном мозге. По данным электроэнцефалограммы невролог диагностирует медленно-волновую активность и наличие мультифокальных спаек.
- Анализы крови. Ценную информацию для постановки диагноза дает исследование уровня церулоплазмина. При синдроме Менкеса, как и болезни Вильсона-Коновалова, этот показатель снижен. Для уточнения диагноза выполняется биохимическое исследование биоптатов печени и обнаруживают уменьшение содержания меди.
- Генетические тесты. Для подтверждения мутации рекомендовано медико-генетическое консультирование ребенка и его родителей. Чтобы выявить патологию гена ATP7A, определяют генетический код и нарушения последовательности ДНК в соответствующем участке Х-хромосомы мальчика. По показаниям проводится обследование матери ребенка.
Лечение болезни Менкеса
Эффективное лечение пока не разработано. Для уменьшения дефицита микроэлементов и улучшения метаболизма медь вводится парентерально в форме гистидината. Такой способ получения минерала гарантирует его доставку ко всем органам и тканям, что активизирует ферментные системы, нормализует окислительное фосфорилирование и работу нервной системы.
Результаты клинических исследований показывают, что раннее назначение меди в парентеральных формах замедляет прогрессирование неврологического дефицита и продлевает жизнь детям с болезнью курчавых волос. У них заболевание протекает в более легкой форме, без судорог и тяжелых когнитивных нарушений. Сложность лечения заключается в своевременной диагностике патологии, что связано с ее редкой встречаемостью.
Симптоматическая терапия включает анальгетики, противосудорожные и психотропные препараты. Они призваны облегчить жизнь пациента. При невозможности естественного кормления вводят зонд либо используют парентеральное питание. Ребенку, у которого выявлена болезнь Менкеса, требуется комплексная реабилитация с участием физиотерапевтов, психологов, логопедов.
Прогноз и профилактика
Учитывая невозможность повлиять на генетические механизмы патологии и отсутствие этиотропного лечение, прогноз неблагоприятный. Большинство детей, страдающих болезнью курчавых волос, живут не более 3 лет. Современные методы реабилитации помогают улучшить качество их жизни и помочь родителям в уходе за особенным малышом. Профилактика синдрома Менкеса включает генетическое консультирование пар с отягощенным семейным анамнезом.

Болезнь Менкеса — это генетический дефект, наследующийся по типу. Впервые патология была описана в 1962 году в работах американского невролога Дж. Менкеса. Данное заболевание характеризуется расстройством транспорта и всасывания меди в кишечнике человека. Возникающая в результате острая нехватка этого элемента в организме препятствует нормальному развитию и функционированию нервной системы, приводит к нарушениям сердечной деятельности, вызывает изменения в структуре кожи, костей сосудов и волос. Заболеванию подвержены только мальчики.
Симптомы заболевания
Болезнь проявляет себя рано, буквально с первых месяцев жизни ребенка. Самыми ранними симптомами обычно бывают:
- частые судороги, порой по несколько раз за день;
- трудности, связанные с кормлением: ребенок вяло и неохотно сосет материнскую грудь, в результате чего с самых первых дней у него наблюдается резкое отставание в росте и весе;
- гипотермия — снижение температуры тела ниже нормы;
- снижение мышечного тонуса;
- задержка умственного развития, тяжелое прогрессирующее поражение мозга;
- гипербилирубинемия — повышенное содержание билирубина в сыворотке крови;
- необычного вида волосы — скрученные и узловатые, чаще всего светлые, словно «выцветшие». Именно этого характерного признака заболевание получило название «Синдром курчавых волос»;
- у некоторых детей наблюдается микрогнатия — недоразвитие верхней челюсти.
Часто малыши с таким диагнозом появляются на свет в результате преждевременных родов. Примерно к трем месяцам симптомы заболевания становятся более очевидными, ребенок утрачивает приобретенные навыки (например, способность держать головку), а лицо малыша приобретает характерные черты: пухлые щечки, как будто «нависающие» челюсти, низкая переносица и изогнутые под углом бровки.
Со временем состояние больного осложняется нарушениями со стороны сосудистой системы, проявляющихся разрывами артерий, кровоизлияниями и тромбозами. Также у больных наблюдаются многочисленные переломы ребер и конечностей, снижение зрения в результате частичного отмирания волокон зрительного нерва, пигментация и гиперэластичность кожи, нарушения со стороны мочеполовой системы.
Существуют данные о мягкой форме болезни Менкеса, которая встречается крайне редко. Для нее также характерно наличие трудностей с кормлением, но при этом отставание по показателям роста весьма незначительно, а задержка психомоторного развития менее выражена. Больные, как правило, также имеют характерные для синдрома Менкеса курчавые и тусклые волосы, однако эти проявления выражены гораздо слабее.
Одной из разновидностей мягкой формы болезни Менкеса является «синдром затылочных рогов», основной признак которого — поражение соединительной ткани. Рентгенографическое исследование скелета показывает его деформацию вследствие разрастания костной ткани. Умственное развитие таких пациентов находится в пределах нормы, правда, на нижней ее границе. Заболевание начинает развиваться в возрасте до 10 лет, что несколько выше, чем при классической форме болезни Менкеса.
Способы диагностики синдрома Менекса
Следует отметить, что все редкие болезни затруднительно диагностировать плохой осведомленности детских врачей о существовании данных заболеваний. В случае болезни Менкеса внимание родителей и педиатра в первую очередь может привлечь необычный вид волос ребенка, а уже микроскопическое исследование помогает определить особенности структуры волоса, характерные для синдрома Менкеса. Весьма показательным симптомом болезни Менкеса являются часто повторяющиеся судороги, постоянное нарастание тяжести данного симптома и отсутствие адекватной реакции на обычное противосудорожное лечение.
Для подтверждения диагноза обычно проводят компьютерную и томографию, компьютерную электроэнцефалографию, рентгенографию трубчатых костей, определение уровня меди и церулоплазмина в сыворотке крови.

Звоните: +7 (495) 222-13-94
Лечение синдрома Менекса
Синдром Менкеса носит прогрессирующий характер и имеет очень тяжелый прогноз. Данное заболевание является неизлечимым, как и остальные редкие генетические болезни. Как правило, больные умирают в первые три года жизни в результате инфекции, пневмонии или разрыва крупных вен. На сегодняшний день лечение состоит во внутривенном введении препаратов меди. Данная терапия не позволяет полностью излечить заболевание: лишь при условии начала лечения с самых первых недель жизни до наступления тяжелых повреждений мозга можно добиться некоторого облегчения течения болезни и незначительного увеличения продолжительности жизни маленького пациента. В остальных случаях лечение неэффективно.
Девочки! Давайте делать репосты.
Благодаря этому к нам заглядывают специалисты и дают ответы на наши вопросы!
А еще, вы можете задать свой вопрос ниже. Такие как вы или специалисты дадут ответ.
Спасибки ;-)
Всем здоровых малышей!
Пс. Мальчиков это тоже касается! Просто девочек тут больше ;-)

Содержание:
- Определение
- Причины
- Симптомы
- Диагностика
- Профилактика
Определение
Болезнь Менкеса (Синдром Менкеса, Трихополиодистрофия) - наследственное генетическое заболевание, из-за аномального гена, ATP7A.Он вызывает нарушение всасывания меди. Синдром Менкеса синдром встречается редко: 1 из каждых 50000-100000 новорожденных.
Причины
Наследование этого синдрома определяется сцепленным с полом рецессивным геном. Это состояние было впервые описано лишь в 1962 г., но удалось установить, что его частота составляет один случай на 3500 живых новорожденных.
Частичная блокада всасывания меди в кишечнике приводит к выраженному дефициту меди, с которым связывают возникновение патологических изменений.
Анализ измененных волос выявляет девятикратное увеличение содержания свободных сульфгидрильных групп по сравнению со здоровым контролем. Аналогичные, но менее выраженные изменения были обнаружены в шерсти овец при дефиците меди.
Патоморфологическая картина. Внутренняя эластическая пластинка артерий фрагментируется, что приводит к появлению извитости артерий и широким колебаниям в их диаметре. В мозговой ткани отмечается глиоз и кистозная дегенерация. В метафизах длинных костей обнаруживаются изменения, напоминающие таковые при цинге.
Симптомы
Волосы.
В момент рождения волосы не изменены. По мере выпадения этих волос они замещаются короткими, ломкими, светлого цвета курчавыми волосами, которые при микроскопическом исследовании обладают признаками перекрученных волос. В первые недели жизни изменены лишь немногие волосы. Отмечалась также узловатая трихоклазия, но это неспецифическая аномалия, которая часто наблюдается в стержнях волос, имеющих дефекты структуры. В наблюдениях отмечен монилетрикс, но обследование большого числа больных из нескольких семей выявило лишь «перекрученные волосы».
Кожа.
Обычно кожа бледная; особенно сильно бледность кожи была выражена у одного ребенка из негритянской семьи. Лицо имеет отличительные черты; щеки пухлые, выражение лица лишено эмоциональной подвижности, брови горизонтальные, волосы на них перекручены.
Системные изменения.
В течение первых 2 мес. ребенок может выглядеть внешне совершенно здоровым. Прогрессирующее замедление психики и двигательной активности начинается с 3-го месяца, когда ребенок становится сонливым и вялым. Нарушается терморегуляция, отмечается высокая чувствительность к инфекции. Часто имеют место судороги, обычно в виде миоклонических поддергиваний. Ребенок, как правило, не переживает 1-2 лет.
Диагностика

До появления характерных изменений волос в возрасте около 3 мес. диагноз может быть заподозрен на основании системных симптомов и выражения лица. Подозрение может быть подкреплено рентгенологическим исследованием и подтверждено определением уровня меди в сыворотке крови.
Гетерозиготные. Облигатные гетерозиготные лица могут иметь перекрученные волосы, но волосы у них могут быть и нормальными. Уровень меди в сыворотке крови в норме. В фибробластах кожи гетерозиготных особей в первичной культуре отмечается метахромазия. Этот тест может оказаться полезным в выявлении носителей среди родственников женского пола. В сыворотке крови снижены уровни меди и церулоплазмина. Уровень меди снижен и в волосах.
Профилактика
При накоплении подробных сведений о характере метаболических дефектов возможным методом лечения может стать парентеральное введение меди. Раннее лечение включает подкожную или внутривенную инъекции с добавкой меди (в форме ацетата соли). Другое лечение симптоматическое и поддерживающее.

Патологическое состояние, характеризующееся нарушением клеточной транспортировки меди, сопровождающееся замедлением роста ребенка и развитием поражений нервной системы. Для заболевания характерно закручивание волос, вследствие чего его также называют болезнью курчавых волос.
Причины
К группе заболеваний, которые возникают на фоне дефицита транспортных каналов меди, относят классическую болезнь Менкеса, мягкий вариант болезни Менкеса и синдром затылочного рога.
Медь участвует в человеческом организме в функционировании самых разных ферментных систем. При дефиците меди у плода могут формироваться различные пороки развития и стигмы дисэмбриогенеза. В результате нарушения свойств эластина и коллагена снижается резистентность сосудистой стенки, что приводит к кровоизлияниям и нарушениям васкуляризации органов. При нехватке этого элемента может отмечаться рождение недоношенных детей, которые в раннем неонатальном периоде часто страдают гипотермией и непрямой гипербилирубинемией. у новорожденных с данной патологией отмечается развитие характерных черт лица, таких как толстые нависающие челюсти, раздутые щеки, аномальные волосы и брови, гипермобильность суставов, дивертикулы мочевого пузыря и уретры.
Заболевание возникает в результате мутации гена ATP7A, который отвечает за кодировку одного из полипептидов трансмембанного белка, в результате чего происходит нарушение транспорта ионов меди. Патогенетические механизмы, которые вызывают заболевание, обусловлены сочетанной недостаточностью медьсодержащих белков, участвующих в окислительном фосфорилировании, продукции меланина, выработке коллагена и кератина, дезинтоксикации свободных радикалов и секреции дофамина.
Симптомы
В большинстве случаев патология манифестирует в неонатальный период. К числу ее ранних клинических проявлений относят гипотеpмию, гипеpбилиpубинемию, задержку физического развития. В неонатальный период у ребенка волосы, как правило, не изменены, но у примерно половине больных встречается образование узелков на волосах и веретенообразные волосы. Довольно часто у таких пациентов отмечается развитие себорейного дерматита. Примерно к 2 или 3 месяцам у ребенка прослеживается отставание роста, задержка психомоторного развития и появление прогрессирующих неврологических расстройств с потерей ранее приобретенных навыков. У больных возникают различные типы эпилептических приступов. По мере развития патологии отмечается возникновение спастического тетрапареза. С течением времени трихополидистрофия становится более очевидной, так как у больного отмечается спутанность волос, они тусклые и не имеют характерного блеска, помимо этого волосы у таких больных седые или имеют цвет слоновой кости и являются достаточно жесткими на ощупь.У некоторых больных появляются нарушения в работе сосудистой системы в виде субдуральных кровоизлияний, разрывов артерий и тромботической болезни. При ангиографии выявляется удлинение сосудов в головном мозге, внутренних органах и конечностях. При контрастной рентгенографии обнаруживаются дивертикулы мочевого тракта, которые могут привести к разрывам и предрасполагают к рекуррентным инфекциям мочевого тракта, что может создать множество проблем пациентам с более длительным сроком жизни. Помимо этого, у больных часто возникают патологические переломы ребер, гиперрастяжимость кожи, диффузная гипопигментация кожи, гиперподвижность суставов.
Диагностика
Постановка диагноза происходит на основании тщательного обследования, сбора анамнеза и данных, полученных при проведении лабораторно-диагностических исследований. Для подтверждения диагноза больному может потребоваться проведение магниторезонансной томографии головного мозга и электроэнцефалограммы головного мозга. Наиболее информативным методом при данной патологии является определение уровня меди и церулоплазмина в сыворотке крови.
Лечение
Больным с данной патологией показано введение гистидината меди в дозировке от 0,2 до 0,45 мг/день внутривенно, что обеспечивает восстановление нормального содержания ионов меди и медь-зависимых белков в сыворотке, печени и цереброспинальной жидкости. При данном заболевании лечение тяжелых неврологических нарушений малоэффективно. Для поддержания организма таким больным могут быть назначены витаминотерапия, основанная на преимущественном введении витаминов группы В.
Профилактика
На данный момент методы, позволяющие снизить вероятность развития данной патологии не разработаны, однако лицам с неблагоприятным семейным анамнезом при планировании зачатия рекомендуется посетить генетика.
Читайте также: